
Orally Dissolving Films (ODFs)
publisherSteve
time2020/04/10

- The manufacture of orally dissolving films is done by various methods such as solvent casting, hot-melt extrusion, semisolid casting, solid-dispersion extrusion, and rolling. The authors discuss these methods and the various parameters in which dissolving films are evaluated.
The manufacture of orally dissolving films is done by various methods such as solvent casting, hot-melt extrusion, semisolid casting, solid-dispersion extrusion, and rolling. The authors discuss these methods and the various parameters in which dissolving films are evaluated.
Jan 02, 2011
By Pharmaceutical Technology Editors
Pharmaceutical Technology
Volume 35, Issue 1
Oral thin films or orally dissolving films (ODFs) provide quick release of an active pharmaceutical ingredient (API) when placed on the tongue. ODFs provide an alternative to orally disintegrating tablets. These dosage forms are placed on a patient's tongue or any oral mucosal tissue. When wet by saliva, the film rapidly hydrates and adheres onto the site of application. It rapidly disintegrates and dissolves to release the medicine for mucosal absorption or, with modifications, allows for oral gastrointestinal absorption with quick-dissolving properties. These films initially were launched as mouth-freshening products containing ingredients such as menthol and thymol. These films are available as breath-freshening products from Johnson & Johnson (New Brunswick, NJ) and Wrigley (Chicago) in the United States and Europe and Boots (Nottingham) in the United Kingdom. Zengen (Woodland Hills, CA) produces a chloraseptic relief strip in the US to deliver benzocaine, a local anaesthetic to treat sore throats.
These ODFs contain film-forming polymers such as hydroxypropylmethyl cellulose (HPMC), hydroxypropyl cellulose (HPC), pullulan, carboxymethyl cellulose (CMC), pectin, starch, polyvinyl acetate (PVA), and sodium alginate. Additional ingredients that are incorporated include plasticizers, sweetening and flavoring agents, coloring agents, saliva-stimulating agents, and thickening agents. Suitable uses for rapidly dissolving films are nicotine-replacement transdermal delivery, and as antiulcer and antihistamine drugs. Antipsychotic and sleeping-disorder drugs also are potential candidates for prescription products (1–4). Advantages of ODFs include improved portability, ease of administration, accurate dosing, cost-effectiveness, and improved patient compliance.
Manufacture of ODFs
One or a combination of the following process can be used in the manufacturing of ODFs: solvent casting, semisolid casting, hot-melt extrusion (HME), solid-dispersion extrusion, and rolling (1, 4). The most commonly used methods of film manufacturing are solvent casting and HME.
Solvent-casting method. The ODF is preferably formulated using the solvent-casting method, whereby the water-soluble ingredients are dissolved to form a clear, viscous solution. The API and other agents are dissolved in smaller amounts in the solution, and combined with the bulk drug. This mixture is added to the aqueous, viscous solution. The entrapped air is removed by vacuum. Deaeration is necessary to obtain uniform film property and thickness. The resulting solution is cast as a film, allowed to dry, and cut into pieces to the desired size. The properties of the API play a critical role in the selection of a suitable solvent. The physicochemical properties of the API should be considered. These properties include compatibility of the API with other film-forming excipients, compatibility with solvents, the polymorphic nature of the API selected, and temperature sensitivity. Manufacturing and packaging ODFs requires special precaution to be taken to control the effect of moisture. Figure 1 indicates critical factors involved in ODF manufacture using the solvent-casting method. Stability of the film and its mechanical properties are significantly affected by the presence of moisture. Another factor requiring strict control is temperature. Controlled temperature conditions are required for maintaining the viscosity of the solution and temperature sensitivity of the API (4).
Specific types of equipment such as rollers are required for pouring the solution on an inert base. The clearance between the roller and the substrate determines the required thickness of the film. The final step, drying the film, removes the solvent and helps to obtain the finished product. Usually, glass, plastic, or teflon plates are used as an inert base for film casting. When the manufacturing technology is transferred from laboratory scale to production scale, several problems can be encountered. These problems can include the casting of the film, obtaining uniform thickness of the film, and proper drying of the sample. The selection of the proper type of dryer is needed in the final step of drying.
Once the films are dried, cutting, stripping, and packaging is done. Suitable size and shapes of films can be cut. The commonly available sizes of films are 3 x 2 cm2 and 2 x 2 cm2. Selection of the packaging container is an equally important parameter for the ODF. The packaging container should provide sufficient mechanical strength to protect the film during shipping and from external factors such as temperature and humidity. Depending upon the characteristics of the film, single-unit containers and multiple-unit dispensers can be selected. The packaged films are inspected before being packed into a secondary packaging container (4).
Hot-melt extrusion . HME is commonly used to prepare granules, sustained-release tablets, and transdermal and transmucosal drug-delivery systems (5). The HME process recently has gained popularity in the pharmaceutical industry. Based on knowledge from the plastics industry, formulators can extrude combinations of drugs, polymers, and plasticizers into various final forms to achieve desired drug-release profiles (5). Processing films by this technique involves shaping a polymer into a film via the heating process rather than through the traditional solvent-casting method (4).
Advantages of HME for film formation include the following:
·No need to use solvent or water
·Fewer processing steps
·Compressibility properties of the API may not be of importance
·Good dispersion mechanism for poorly soluble drugs
·More uniform dispersion of the fine particles because of intense mixing and agitation
·Less energy compared with high-shear methods
·Minimum product waste
·Possibility of scale-up
·Good control of operating parameters.
In the HME process, the API and other excipients are mixed in a dry state, the heating process is started, and the molten mass is extruded out of the hot-melt extruder. The advantage of this process is the complete elimination of the solvent. The films are allowed to cool and are cut to the desired size. The high temperature used in the process makes it suitable for thermostable drugs. Drugs that are sensitive to temperature cannot be used in this process.
Table I compares solvent-casting and HME for the manufacture of ODFs. Solvent casting is a hydrous process suitable for thermolabile and thermostable drugs in comparison to HME, which is anhydrous and requires thermostable drugs. Repka et al. studied the influence of chlorpheniramine maleate (CPM) on topical HPC films by HME (5). CPM has been reported to function as an effective plasticizer, thereby increasing percent elongation and decreasing tensile strength in a concentration-dependent manner. CPM also acts as a processing aid in the extrusion of hot-melt films by allowing film processing at lower temperatures (6).
An evaluation of HME and the in vivo bioadhesive properties of HPC films containing seven polymer additives on the epidermis of human subjects was performed (7). HPC films containing additives with and without plasticizers were prepared by HME. Incorporation of a carbomer (Carbopol 971P NF, Lubrizol, Cleveland, OH) and polycarbophil into HPC films increased bioadhesion significantly. Many studies were conducted using HME for preparing solid dispersions. It was reported that melt extrusion of miscible components resulted in amorphous solid-solution formation, whereas extrusion of an immiscible component led to the amorphous drug dispersed in a crystalline excipient (8). The process has been useful in preparing solid dispersions in a single step. An extruder consists of two distinct parts. The first part consists of a conveyer system that transports the material and imparts a degree of distributive mixing. A second part, a dye system, forms the materials into the required shape. The drug-carrier mix is filled in the hopper and is conveyed, mixed, and melted by the extruder. The die shapes the melt in the required form such as granules, pellets, films, or powder, which can be further processed into conventional tablets or capsules. Oxygen and moisture should be completely eliminated for substances susceptible to oxidation and hydrolysis (9).
Semisolid casting . In the semisolid-casting method, a solution of the water-soluble, film-forming polymer is prepared. The resulting solution is added to a solution of acid insoluble polymer (e.g., cellulose acetate phthalate and cellulose acetate butyrate), which is previously prepared in ammonium or sodium hydroxide. The appropriate amount of plasticizer is added to obtain a gel mass. The prepared gel mass is cast into films or ribbons using a controlled heat source. The thickness of the film is controlled between 0.015–0.05 in. (9).
Solid-dispersion extrusion . The term solid dispersion refers to the dispersion of one or more APIs in an inert carrier in a solid state in the presence of amorphous hydrophilic polymers using methods such as HME. In solid-dispersion extrusion, immiscible components are extruded with drug, and solid dispersions are prepared. The solid dispersions are shaped into films by means of dies. The drug is dissolved in a suitable liquid solvent. This solution is incorporated into the melt of polyols such as polyethylene glycol, obtained below 70 °C, without removing the liquid solvent. The selected solvent or dissolved drug may not be miscible with the melt of the polyethylene glycol. The polymorphic form of the drug precipitated in the solid dispersion may be affected by the liquid solvent used (9, 10).
Rolling method . In the rolling method, a solution or suspension containing the drug is rolled on a carrier. The solvent is mainly water and a mixture of water and alcohol. The film is dried on the rollers and cut into desired size and shapes. The film is made by preparing a premix and adding the API, and film is subsequently formed (11). The premix or master batch containing the film-forming polymer, polar solvent, and other excipients, except the API, are added to the master-batch feed tank. A predetermined amount of the master batch is controlled and fed through a metering pump and control valve to the mixers. The required amount of the drug is added to the desired mixer through an opening. After blending the API with the master batch to provide a uniform matrix, the matrix is fed to the pan using metering pumps. The thickness of the film is controlled using a metering roller. The film is finally formed on the substrate and carried away via the support roller. The wet film is dried using controlled bottom drying, preferably in the absence of external air currents or heat on the surface of the film.
Evaluation of the ODF
The ODF is evaluated by various parameters such as thickness, the mechanical properties of the film, folding endurance, assay/drug content as well as by studies of in-vitro disintegration, in-vitro dissolution, surface morphology, and taste (12, 13).
Thickness. The thickness of strip can be measured by a micrometer at different locations. This measurement is essential to ascertain uniformity in the thickness of the film as this thickness is directly related to the accuracy of the dose in the strip.
Mechanical properties of the film . The mechnical propertis are tensile strength, percentage elongation, and elastic modulus.
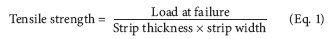
Tensile strength. Tensile strength is the maximum stress applied to a point at which the strip specimen breaks. It is calculated by the applied load at rupture divided by the cross-sectional area of the strip as given in the equation below:
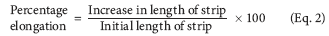
Percentage elongation. When stress is applied, a film sample stretches, and this stress is referred to as strain. Strain is basically the deformation of the film divided by the original dimension of the sample. As the plasticizer content increases, the elongation of film is observed.
Tear resistance. The tear resistance of a plastic film is a complex function of its ultimate resistance to rupture. A very low rate of loading of 51 mm/min is employed. It is designed to measure the force to initiate tearing. The maximum stress or force (usually found near the onset of tearing) required to tear the specimen is recorded as the tear resistance in newtons.
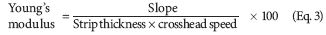
Young's modulus or elastic modulus. Young's modulus or elastic modulus is the measure of the stiffness of the film. It is represented as the ratio of applied stress divided by the strain in the region of elastic deformation:
Hard and brittle strips demonstrate a high tensile strength and Young's modulus with less percentage elongation.
Folding endurance. Folding endurance is determined by repeated folding of the film at the same place until the film breaks. The number of times the film is folded without breaking is calculated as the folding endurance value.
Assay/drug content. Assay/drug content is determined by any standard assay method described for the particular API in any of the standard pharmacopoeia.
In-vitro disintegration. Disintegration time gives an indication about the disintegration characteristics and dissolution characteristics of the film. For this study, the film, as per the dimensions required for dose delivery, was placed on a stainless-steel wire mesh containing 10 mL of distilled water. The time required for the film to break was noted as in-vitro disintegration time.
In-vitro dissolution. In-vitro dissolution studies can be performed using the modifications to the standard basket or paddle apparatus described in any of the pharmacopoeia because a conventional paddle apparatus may lead to floating of the film. The dissolution medium will be selected as per the sink conditions and the highest dose of the API.
Surface morphology . A study of surface morphology of the ODF is performed using the environment-scanning-electron microscopy method. The uniformity of the film and absence of pores and striations indicate the good quality of the ODF.
Taste evaluation. A taste evaluation study can be performed using a panel of human volunteers. The ODF should possess the desired sweetness and flavor acceptable to the patient. In-vitro methods using taste sensors, a specially designed apparatus, and drug release by modified pharmacopoeial methods are used for this purpose. Experiments using electronic-tongue measurements also have been reported to distinguish between the sweetness levels in taste-masking formulation.
Clinical and regulatory requirement
To indicate bioequivalency of a product to that of existing oral drug, an abbreviated new drug application is required. In-vitro dissolution studies and therapeutic equivalence are considered. Comparative bioequivalence between an orally disintegrating tablet and an ODF can be evaluated. If the ODF exhibits a different target pharmacokinetic profile compared with the existing marketed product, the ODF is considered a new dosage form. For a new dosage form, a new clinical study is required. A new clinical study offers the advantage of three years of marketing exclusivity to the product. Preclinical toxicity studies are not required if the molecule is the same as that of the approved product. Safety, tolerability, and efficacy features are to be demonstrated in such trials. Oral mucosa-irritation testing is carried out in both animal models and humans. The hamster-cheek pouch is the most appropriate model for predicting irritation criteria before testing in humans (12).
Conclusion
ODFs are a possible alternative dosage form to orally disintegrating tablets. These films offer the benefits of pleasant mouthfeel and rapid disintegration in the mouth. Solvent casting, hot-melt extrusion, semisolid casting, solid-dispersion extrusion, and rolling are important manufacturing methods to produce these films.
Renuka Mishra* is an assistant professor, and Avani Amin is a professor in the Department of Pharmaceutics and Pharmaceutical Technology, the Institute of Pharmacy, Nirma University, Ahmedabad, Gujarat, India, Sarkhej-Gandhinagar Highway, Ahmedabad, Gujarat, India, renukasharma81..........com
*To whom all correspondence should be addressed.
References
1. S. Borsadia, D. O'Halloran, and J.L. Osborne, Drug Del. Tech. 3 (3), 63–66 (2003).
2. T. Ghosh and W.Pfister, "Intraoral Delivery Systems: An Overview, Current Status and Future Trends," in Drug Delivery to the Oral Cavity: Molecules to Market, T. Ghosh and W. Pfister, Eds. (Taylor & Francis, Florida, CRC Press, 2005), pp. 1–34.
3. P. V. Arnum, "Outsourcing Solid Dosage Manufacturing," Pharm. Technol. 30 (6), 44–52 (2006).
4. R. Mishra and A. Amin, Pharm. Technol. Eur. 19 (10), 35–39 (2007).
5. M. Repka et al., "Hot Melt Extrusion," in Encyclopedia of Pharmaceutical Technology, J. Swarbrick and J. Boylan, Eds. (Marcel Dekker Inc., New York, Vol. 2, 2nd Edition, 2002), pp. 1488–1504.
6. M.A. Repka and J.W. McGinity, Pharm. Dev. Technol. 6 (3), 297–304 (2001).
7. M. Repka and J.W. McGinity, J. Controlled Release 76 (3), 341–351 (2001).
8. J. Breitenbach, Eur. J. Pharm. Biopharm. 54 (2), 107–117 (2002).
9. A. Arya et al., Int. J. Chem. Tech. Research 2 (1), 578–583 (2010).
10. Gole et al., "Pharmaceutical and Other Dosage Forms," US Patent 5648093, Jul. 1997.
11. R.K. Yang et al., "Thin Film with Non-Self Aggregating Uniform Heterogeneity and Drug Delivery Systems Made Therefrom," US Patent Application 20080226695.
12. R. Mishra and A. Amin, Pharm. Technol. 33 (2), 48–56 (2009).
13. R.P. Dixit, S.P. Puthli, J. Controlled Release 139 (2), 94–107 (2009).